

Blogpost 1 – An unexpected impurity in my drug product, what now?
6-7 min reading time
Welcome to the first article in a series of blog posts on impurities in drug products. These blog posts will focus on different situations where unexpected drug impurities are detected in a quality control analysis. All cases that will be presented have been structure elucidation projects at Nelson Labs, hence real-world examples from pharmaceutical or medical device industries. The identification of an unknown impurity is key in the qualification process, so for each case the analytical strategy will be briefly discussed. This first blog is an introduction on how to handle (unexpected) impurities in a drug substance or drug product.
What are organic drug impurities in the first place?
Generally speaking, impurities are chemical substances that are unintentionally present in a drug substance or product. Impurities do not contribute to the intended therapeutic effect. On the contrary, they may adversely affect the drug product’s efficacy and safety. More formally, a drug substance impurity is defined by the ICH Q3A (R2) & ICH Q3B (R2) guidelines as “any component that is not the chemical entity defined as the drug substance.” Similarly, a drug product impurity is “any component of the new drug product that is not the drug substance or an excipient in the drug product” (ref. 1-2).
How can we classify organic drug impurities?
Based on their origin, three main categories of organic impurities can be distinguished:
- Process-related impurities: associated to the synthesis of the drug substance
- Degradation impurities: associated to the stability of the active pharmaceutical ingredient (API)
- Contamination impurities: impurities that are not directly drug related
A detailed classification of pharmaceutical impurities and their origin is presented in Figure 1.
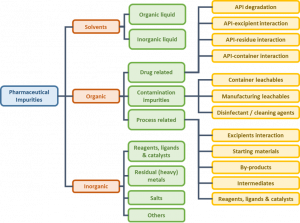
Figure 1: Classification of impurities (ref. 3-4)
Where do impurities come from?
The blog posts will mainly focus on organic impurities and residual solvents. During drug development, much effort is put into characterization of process-related impurities and degradation impurities. After establishing the impurity profile of the drug substance, the synthesis process is controlled by various measures such as incoming inspection of raw materials, in-process controls, compendial tests etc. Unexpected impurities from the synthesis process should be rare but may occur when the synthesis is poorly understood, a procedure was changed without appropriate change control, or a raw material supplier altered the manufacturing of a raw material without further notice. Drug-related degradation impurities are thoroughly investigated by means of stability & forced degradation studies, hereby stressing the drug product at exaggerated conditions to profile as much as possible API degradation impurities. Such studies are inherently designed to prevent the occurrence of unknown degradation impurities in future drug batches intended for market release.
As upcoming blog posts will reveal, unexpected impurities are often contamination impurities such as leachables or residual contaminants from manufacturing or cleaning. Leachables are characterized by means of leachables studies. In these studies, the drug product is brought into contact with the container closure system and the substances that migrate from the packaging into the drug product are analyzed. Unexpected leachables can occur when leachables react with the drug substance or formulation components or changes are made to the packaging or manufacturing systems where new leachables are encountered.
How to deal with impurities?
The ICH Q3A & ICH Q3B guidelines have established reporting, identification, and qualification thresholds, based on estimated patient exposure. Once the impurity’s level in the drug product breaches the reporting threshold, the impurity is reported, which is no more rigorous than noting “there is an impurity above the reporting threshold.” In this circumstance, no additional actions are required unless the impurity’s level breaches the identification threshold. When this occurs, both the existence and the identity of the impurity must be documented (reported), meaning that the identity must be ascertained. In this case, no further toxicological assessment is required, although the identity is typically examined to establish whether the impurity is “unusually potent.” Should the impurity’s level increase sufficiently so that it breaches the qualification threshold, the reported and identified impurity must be qualified, meaning that you must establish the impurity’s biological safety. For the qualification of unexpected impurities, securing their identification is perhaps the most critical step as it is a compound’s identity that is linked to its functionality, and it is its functionality that dictates its toxicity.
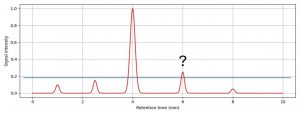
Figure 2: Impurity peaks in the chromatogram above a particular threshold need to be reported, identified or qualified.
How to identify drug impurities?
Organic impurities are usually detected with a chromatographic technique such as gas chromatography (GC) or liquid chromatography (LC). The identity may be revealed from the information present in the chromatographic peak, that is the retention time and in some cases the detector response might contain structure-related information. Mass spectrometry (MS) is widely used as a detector that produces a spectral response in function of the chemical structure. However, Quality Control (QC) assays usually do not routinely use MS as standard equipment so spectral information for structure elucidation is lacking when an unexpected impurity is detected for the first time. To identify the impurity, additional analyses are required using analytical techniques that produce spectral information in function of the chemical structure, such as MS or Nuclear Magnetic Resonance (NMR) spectroscopy.
Best practices regarding structure elucidation strategies and associated challenges will be presented in future blog posts through anonymized case studies revealing the analytical strategy used to identify the unexpected drug impurity.
If you have additional questions about Impurities Identification test services or would like to consult with the experts at Nelson Labs, just send an e-mail to [email protected].
References
[1] ICH Q3A (R2) Step 5 – Impurities in new drug substances
[2] ICH Q3B (R2) Step 5 – Impurities in new drug products
[3] R.J. Smith, M.L. Webb; Analysis of Drug Impurities; Blackwell Publishing; 2007
[4] K. Liu, C. Chen; “Determination of Impurities in Pharmaceuticals: Why and How?” In: Quality Management and Quality Control – New Trends and Developments, edited by P. Pereira, S. Xavier; IntechOpen; 2019

